
Pej Lab
The Pej Lab is interested in the design of probabilistic machine learning methods and statistical models that incorporate known biochemical principles to facilitate decision-making from limited data and to allow for asking smarter questions. We have a major focus on quantitative analysis of regulatory variation in the human genome from large scale functional genomics data and its application to rare diseases and precision medicine.
Explore Our Research
We have a long-standing interest in building statistical methods that systematically incorporate biochemical principles or expert knowledge to effectively extract interpretable results from otherwise limited and noisy experimental data. Our lab applies this strategy to developing quantitative models of genetic regulatory variation. Genetic variation in the regulatory genome plays a major role in human phenotypic variation and disease susceptibility. Currently, our ability to interpret regulatory variation in human genome is hampered by over-simplistic models and limited statistical power. Our efforts to address some of the challenges in interpreting regulatory genome fall broadly into the following two categories.
Rare disease diagnostics
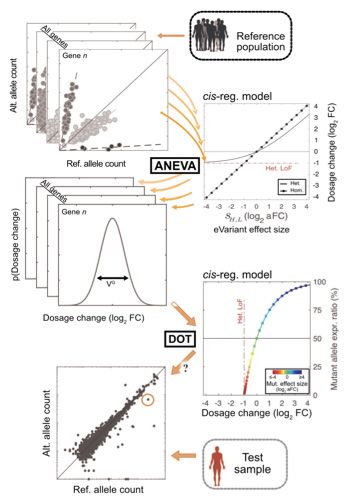 Rare diseases are conditions that affect less than 1 in 2000 people in the population. There are >7000 identified rare diseases affecting up to 30 million Americans. The large majority of rare diseases are genetic and manifest early on in life, often with severe health consequences. In more than 60% of the cases in which a rare genetic disease is suspected, whole-genome sequencing fails to identify coding variants that lead to protein truncation or are otherwise potentially pathogenic. Rare variants that affect gene regulation are expected to underlie pathogenesis in some of these cases.
Rare diseases are conditions that affect less than 1 in 2000 people in the population. There are >7000 identified rare diseases affecting up to 30 million Americans. The large majority of rare diseases are genetic and manifest early on in life, often with severe health consequences. In more than 60% of the cases in which a rare genetic disease is suspected, whole-genome sequencing fails to identify coding variants that lead to protein truncation or are otherwise potentially pathogenic. Rare variants that affect gene regulation are expected to underlie pathogenesis in some of these cases.
We develop new methods that allow for using transcriptome data to increase diagnostic yield for rare disease patients. These include devising appropriate statistical tests for identifying regulatory aberrant genes and developing mathematical models for identifying the most appropriate tissues for transcriptome profiling in each patient. These efforts are in close collaboration with scientists and clinicians at Neuromuscular and Neurogenetic Disorders of Childhood Section at the NIH, Rady Children’s Institute for Genomic Medicine, and the center for Undiagnosed Disease Network in Stanford university. Below are two of our publications in this area.
- Transcriptomic signatures across human tissues identify functional rare genetic variation.
N Ferraro, B Strober, J Einson, NS Abell, F Aguet, …, P Mohammadi†, S Montgomery†, A Battle†.
Science, 2020. - Genetic regulatory variation in populations informs transcriptome analysis in rare disease.
P Mohammadi†, S Castel, B Cummings, J Einson, C Sousa, …, T Lappalainen†.
Science, 2019.
Gene regulation and common disease
Around 90% of genomic loci associated with common diseases, such as cancer, type 2 diabetes, or cardiovascular diseases, fall outside the gene boundaries and are thus hard to interpret. In recent years, there has been a deluge of data from genome-wide functional assays. The rapid expansion of computational techniques for mapping genetic correlates of intermediate molecular traits, such as gene expression, has offered opportunities to explore molecular mechanisms that underlie disease susceptibility. Over the past decade, quantitative trait loci mapping studies have identified tens of thousands of common genetic variants affecting the regulation of virtually every protein-coding gene in the human genome. Various biological aspects of these genetic associations, including genomic context and tissue specificity, disease-modifying effects, contribution to common traits, and the effect of local ancestry are established.
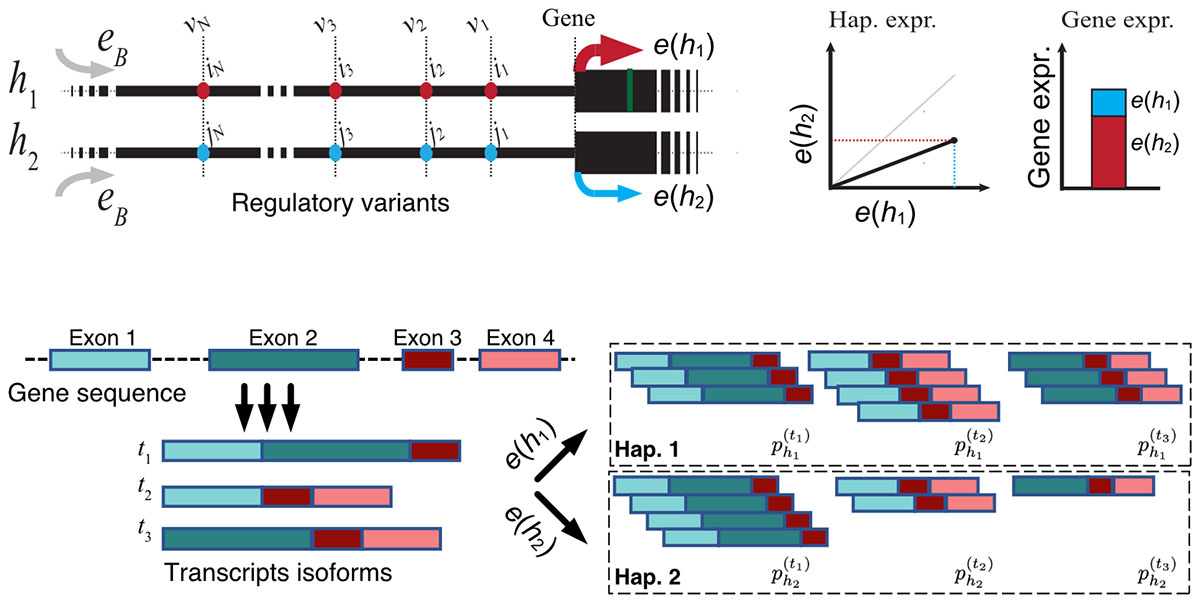
We develop mechanistic models of genetic variation in gene regulation to distill scattered pieces of accumulated knowledge about trends in functional genomic data into unifying theoretical models. These models crystallize our best understanding of the underlying biology and expose the remaining knowledge gaps that remain to be addressed. Using these accurate models of gene regulation, we systematically incorporate the dosage modifying effect of regulatory alleles into genetic association analyses to enhance the resolution of the current genotype-phenotype maps to allow for a more refined mapping of the underlying biological mechanisms that are more generalizable across diverse populations. These efforts are in close collaboration with other scientists in the GTEx consortium and TOPMed project and involve large-scale biobank data from Vanderbilt university bioVU and the UK Biobank. We also contribute to functional analyses of gene expression data (RatGTEx.org) at the national center of excellence for GWAS in Outbred Rats at UC San Diego. Below are a few of our publications in these areas.
- Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change.
P Mohammadi, SE Castel, AA Brown, T Lappalainen.
Genome Research, 2017. - Multimodal analysis of RNA sequencing data powers discovery of complex trait genetics.
D Munro, N Ehsan, SM Esmaeili-Fard, A Gusev, AA Palmer, P Mohammadi.
Nature Communications, 2024. - Haplotype-aware modeling of cis-regulatory effects highlights the gaps remaining in eQTL data.
N Ehsan, Bence M Kotis, SE Castel, EJ Song, N Mancuso, P Mohammadi.
Nature Communications, 2024.

Pejman Mohammadi, PhD
Pejman is a computational biologist with a background primarily in statistical machine learning and Bayesian data modeling. His current research interests span over practical and theoretical issues that arise in genomics and personalized medicine, focusing on quantitative analysis of regulatory variation in the genome and its application to rare diseases.
Pejman jointed UW as an Associate Professor in 2023. Prior to that he was at Scripps Research in San Diego as a tenure track Assistant Professor in 2018, later as an Associate Professor from 2021. He holds a Ph.D. in Computational Biology from ETH Zurich and carried out postdoctoral work at the NY Genome Center and Columbia University from 2015 to 2017.
-

Katharine Chen
Postdoc
Katharine joined the lab as a postdoc in 2024. She is interested in applying and developing computational models to better understand the effects of eQTLs on their target genes using GTEx and other transcriptomic datasets. She holds a PhD in Molecular and Cellular Biology with a specialization in Data Science from the University of Washington, where she worked on developing massively parallel sequencing assays and CRISPR screens for identifying regulators of mRNA translation.
-

Amy Crowson
Laboratory Administrative Coordinator
-

Mehdi Esmaeili-Fard
Postdoc
Mehdi joined the lab in 2023. He is a computational and quantitative biologist and applies statistical methods to detect genomic variations underlying disease/traits. Mehdi also has wide experience in whole-genome prediction using statistical approaches. At the PejLab, Mehdi's work focuses on applying bioinformatics and computational methods to conduct GWAS and eQTL/single-cell eQTL analyses to identify genes and pathways in the cochlear-vestibular system related to balance in older adults. Mehdi is interested in multi-omics analyses using different layers of genomic data that can lead to a better connection between genotype and phenotype and a better systematic understanding of the information flow across different omics layers. "By the way, our ultimate goal is improving human health :)".
-

Kaushik Ram Ganapathy
Graduate Student (PhD Candidate)
Kaushik joined the lab in 2021 and is a second-year graduate student. His thesis project involves developing methods for rare variant outlier detection. He is interested in using data science to help improve human health. Before joining Scripps, he co-founded and led GeoACT, a project to model COVID-19 spread in schools working at the San Diego Supercomputer Center with Dr. Ilya Zaslavsky. Kaushik holds a B.S. in Data Science from the Halicioglu Data Science Institute, UC San Diego.
-

Congyu Hang
Graduate Student
Congyu Hang holds a B.S. in Statistics Specialist Quantitative Finance from University of Toronto. She is currently pursuing her master of Biostatistics at University of Washington. Her current project is about the application of machine learning methods for modeling ASE data to uncover latent regulatory variation in gene expression.
-

Yan Hao
Staff Scientist
Yan joined the lab as a staff scientist in 2024, focusing on the application of statistical machine learning models to analyze single-cell spatial transcriptomics and genomics within clinical trial datasets. Prior to this role, she concentrated her research on functional genomics, aiming to discover new drug targets for neurological disorders, immune diseases, and cancers. Yan holds a PhD in Cellular Biology from the University of Michigan and a Master's degree in Computer Science from Georgia Tech. Her postdoctoral work was conducted at AbbVie.
-

Daniel Munro
Staff Scientist
Daniel joined the lab in 2020 and has a joint position in the Palmer Lab at UC San Diego. He is developing methods to identify regulatory variation in outbred rats with applications in psychiatry, as well as tools for extracting biological features from RNA-Seq. He did his PhD in Quantitative and Computational Biology at Princeton University with Mona Singh.
-

Sarah Silverstein
Graduate Student
Sarah is a fourth-year medical student at Rutgers New Jersey Medical School and current PhD student with the Rutgers-NIH graduate partnership program. She joined the lab of Carsten G Bönnemann MD at the NNDCS/NIH and David Adams MD PhD at the Undiagnosed Diseased Program/NIH in 2020. Her thesis project focuses on using whole transcriptomic sequencing to improve molecular diagnosis in genome and exome negative rare disease patients. Upon graduation, she plans on pursuing clinical training in pediatric neurology with a focus in neurogenetics.
-

Shiyu Wan
Graduate Student (PhD Candidate)
I am interested in applying deep learning techniques to analyze public health and biological data. Prior to joining the lab, I worked on developing a novel deep learning method and a feature importance test for analyzing complex survival data. I hold a Bachelor of Medical Science in Public Health and a B.A. in Economics from Peking University, as well as an M.S. in Biostatistics from the University of North Carolina at Chapel Hill.
-

Tom Willis
Postdoc
Tom joined the lab as a postdoc in 2025. He is interested in the role of common variants in rare disease and methods which leverage cross-trait sharing of genetic architecture. During his PhD he studied the role of common variants in antibody deficiencies and was supervised by Chris Wallace at the MRC Biostatistics Unit, Cambridge. He is also interested in workflow automation and reproducibility.
-

Michael Yung
Graduate Student (PhD Candidate)
Michael joined the lab in 2024 and is a first-year graduate student. His project will involve the analysis of genetic determinants of regulatory variation in humans using previously collected and publicly available RNA sequencing data. He is interested in using statistical learning and high-dimensional data to solve complex problems in genetics and genomics. Before joining the lab, he was working with Bruce Weir on a research project related to the interpretation of Y-STR evidence, particularly on estimating population-specific values of theta for Y-STR Profiles. Michael holds a B.S. in Statistics and B.S. in informatics from University of Washington.