
Hahn Lab
Advanced Proteomics for Newborn Screening and Diagnosis
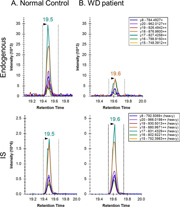
Figure 1. Extracted ion chromatograms for ATP7B 1056 peptide after peptide capture in DBS from (A) normal control and (B) WD patient.
The Hahn laboratory focuses on development and validation of assays that have important clinical applications for population screening, diagnosis and prognosis.
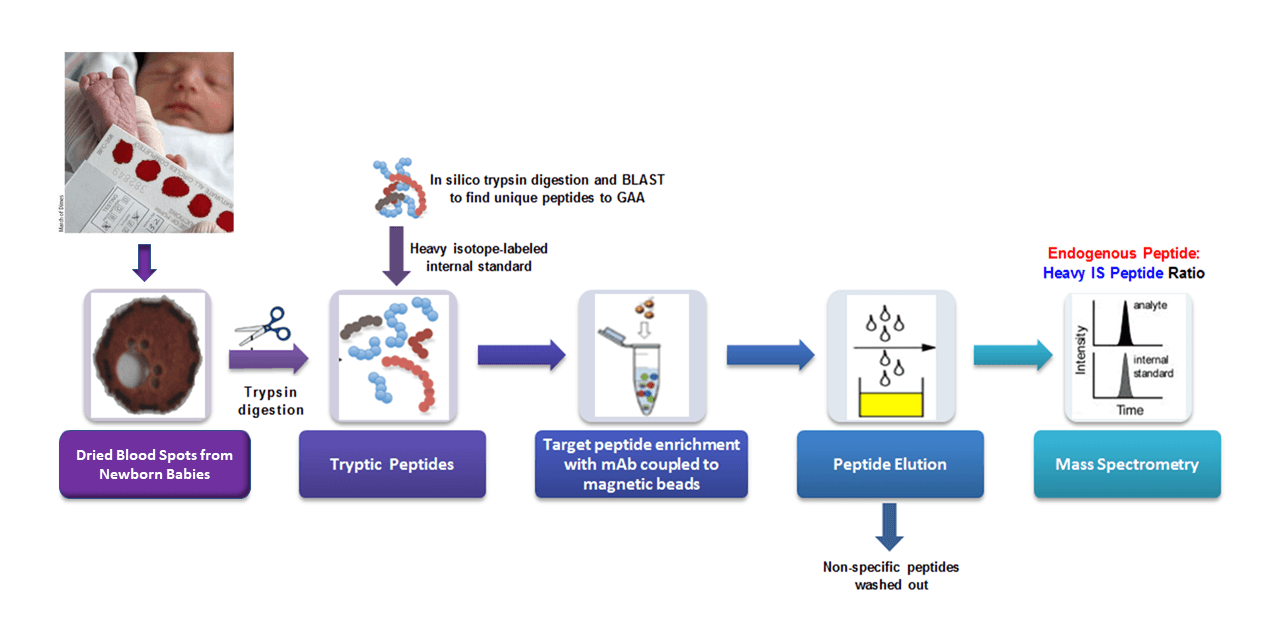
The Hahn Lab is pioneering the use of targeted proteomics for applications in newborn screening and diagnostics from dried blood spots (DBS). We believe that the direct quantification of aberrantly produced proteins by mass spectrometry has great potential for diagnosing genetic disorders which feature reduced, absent or otherwise altered levels of specific target proteins (Jung et al., 2017; deWilde et al., 2008; Kerfoot et al., 2012). The Hahn laboratory has adapted an approach called “Immuno-SRM”, using target-specific monoclonal antibodies to enrich peptide biomarkers from complex matrices, to quantify protein concentrations in DBS (Jung et al., 2017). Signature peptides of a target protein are selectively enriched and quantified by tandem mass spectrometry which allows the diagnosis of a patient with absent or reduced protein of interest. This powerful approach enables the direct detection of extremely low abundance peptides that would have been previously unusable as diagnostic biomarkers.
As a proof of concept, our lab has applied the use of Immuno-SRM screening to Wilson’s Disease (WD), a genetic copper transport disorder where treatments exist but well-validated screens are lacking. The Hahn lab has demonstrated the immuno-SRM method is able to quantify the concentration of ATP7B protein, the membrane transporter responsible for WD, in DBS and utilize it to distinguish the WD patient from healthy individuals as shown in Figure 1 (Jung et al., 2017). To our knowledge, the concentration of ATP7B in DBS of WD patients was always reduced, or absent, when compared to healthy controls. Thus, this method has the potential to serve as a tool for newborn screening of WD.
Currently, we are developing a targeted proteomics-based screen for a group of primary immunodeficiency disorders (PIDD). These disorders comprise a group of life-threatening congenital diseases characterized by absent or impaired immune responses. Effective treatments are available but require early diagnosis for optimal clinical outcomes, meaning that effective newborn screening would provide substantial benefit. Using Immuno-SRM and specific peptide biomarkers, we have shown the ability to differentiate and diagnose patients with X-linked agammaglobulinemia (XLA), Wiskott-Aldrich Syndrome (WAS), and Severe Combined Immunodeficiency (SCID) from one another and from unaffected normal control DBS samples within a single mass spectrometry experiment. These methods are robust, precise, and highly-multiplexed to provide rapid screening. Ongoing work is focused on developing and incorporating peptide biomarkers for DOCK8 deficiency, Common Variable Immunodeficiency, Ataxia Telangiectasia, Familial Hemophagcytic Lymphohistocytosis 2, X-linked Lymphoproliferative Syndrome 1, and X-linked Chronic Granulomatous Disease. A single assay with the ability to screen the population for all these diseases will be of tremendous value to the PIDD community.
An additional set of targets include a group of Lysosomal Storage Disorders including Cystinosis, MPS I, and Pompe Disease. Cystinosis is a rare, but life-threatening lysosomal storage disorder. If untreated, infants develop progressive and severe neurological damage, irreversible multi-organ failures including kidney failure, and premature death. Diagnosing cystinosis soon after birth, before the severe signs of disease develop, offers the best chance for successful and effective treatment. Using primary and secondary signature peptide markers indicative of cystinosis (Cystinosin and Sedoheptulokinase) we hope to enable a multiplexed newborn screen for this disorder.
In addition to develop new assays for diagnosis, Hahn lab also devotes to improve the existing assays for newborn screening. In the case of Pompe disease (PD), both screening method and treatment are well established but studies have shown that a successful treatment could be predetermined by the CRIM status, which is an indicator of whether there is any endogenous acid α-glucosidase present, of Pompe patients. Patients with PD who develop sustained high titers of neutralizing antibodies to Enzyme Replacement Therapy (ERT) that render the treatment ineffective usually also have poor clinical outcomes. Immunomodulation for these groups of patients, ideally before ERT begins, would improve treatment efficiency. Unfortunately, the current screening methods are not able to distinguish between the CRIM-negative and CRIM-positive patients. Based on the high sensitivities of the immuno-SRM method achieved on other assays in Hahn lab, we are also optimistic about this method’s ability to diagnose Pompe patients and give them a specific CRIM designation, which will benefit the treatment planning substantially.
We are on the road to eventually screen or diagnose patients with a wide range of disorders, such as primary immunodeficiencies, Wilson disease, cystinosis and several other lysosomal storage diseases (MPS I, Pompe and Cystinosis) in one single assay. These methods are highly robust, multiplexable and allow analysis of proteins from DBS, samples that are highly stable and collected non-invasively. Upon optimization, these methods have the potential to be applied broadly to newborn screening.

Sihoun Hahn, MD, PhD
Sihoun Hahn, MD, PhD, is a principal investigator at Seattle Children’s Research Institute and Professor of Pediatrics and Adjunct Professor of Medicine at the University of Washington School of Medicine. He directs the Seattle Children’s Biochemical Genetics Program and the Biochemical Genetics Laboratory.
Dr. Hahn joined Seattle Children’s in 2006 after six years at the Mayo Clinic, where he served as Professor of Laboratory Medicine and Pathology, Pediatrics, and Medical Genetics, and as Co-Director of the Biochemical Genetics Laboratory.
Dr. Hahn’s career has been devoted to improving early detection and prevention of rare but treatable genetic disorders through innovative laboratory technologies. His work has focused particularly on developing population-based screening methods for Wilson disease, a genetic disorder of copper metabolism that results in toxic accumulation of copper in the liver, brain, and other organs (Collin C et al., 2021; Jung S et al, 2017; Hahn, 2014; Bennett et al., 2013; Bennett and Hahn, 2011; deWilde et al., 2008; Hahn et al., 2002; Horslen and Hahn, 2010; Kroll et al., 2006; Perri et al., 2005).
Building upon this foundational work, Dr. Hahn led the development and clinical validation of a novel targeted proteomic assay that quantifies ATP7B proteotypic peptides in dried blood spots using liquid chromatography–tandem mass spectrometry (LC-MS/MS). This work culminated in a large-scale population pilot screening of over 30,000 newborns (Klippel C et al, 2025).
In 2025, following review of the scientific and clinical evidence, the Washington State Board of Health approved the addition of Wilson disease to the Washington State Newborn Screening (NBS) panel. This milestone represents one of the first successful implementations of a targeted proteomic mass spectrometry assay for Wilson disease within a public health newborn screening program and marks a significant advancement in translational proteomics and preventive medicine.
In addition to Wilson disease, Dr. Hahn’s funded research programs focus on developing multiplex peptide fingerprinting and targeted proteomic assays for a broad range of treatable genetic disorders, including inborn errors of metabolism and primary immunodeficiencies. His work emphasizes reducing false-positive rates, improving analytical specificity, and creating scalable, high-throughput screening platforms suitable for public health implementation. His research has resulted in numerous peer-reviewed publications and national and international collaborations advancing newborn screening science.
Dr. Hahn is deeply committed to integrating laboratory innovation with clinical practice. He remains a strong advocate for prevention, believing that identifying disease and initiating intervention before symptoms appear is far preferable to treating established organ damage.
He serves as a member of the Wilson Disease Association’s Medical Advisory Committee and the Washington State Newborn Screening Advisory Committee. In recognition of his sustained scientific leadership and service to the Wilson disease community, Dr. Hahn received the 2025 Carol A. Terry Service Award from the Wilson Disease Association. Earlier in his career, he was honored with the American College of Medical Genetics Foundation/Luminex Award (2009).
Dr. Hahn continues to advance proteomics-based newborn screening with the goal of expanding early detection of treatable genetic diseases both nationally and globally.