
Current Research at the Rau Lab
SWI/SNF Inhibition to Treat Pediatric Leukemias
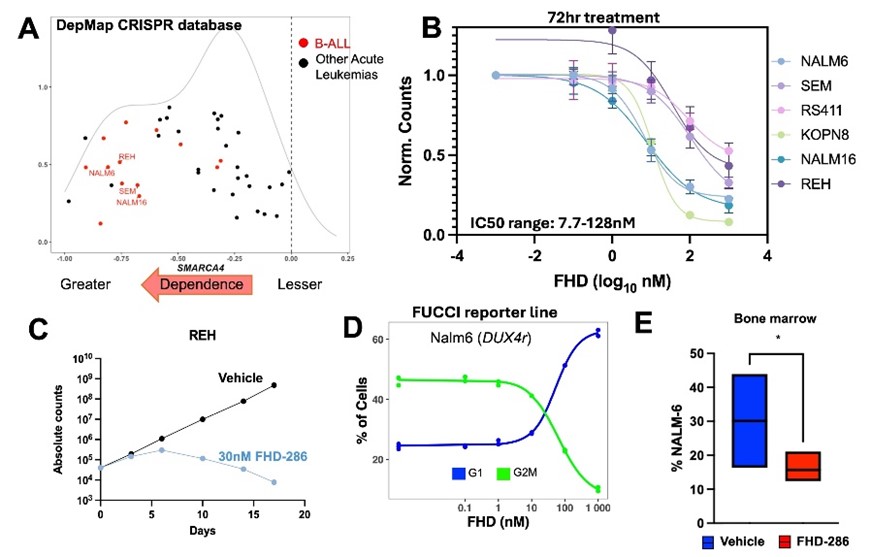
Figure 1: SWI/SNF in B-ALL. A) DepMap dependency scores of leukemia cell lines including B-ALL (red dots). B) Dose response curves of B-ALL cell lines treated with FHD-286 x 72hrs. C) Response of REH cells after prolonged treatment with sub-IC50 dosing. D) Cell cycle analysis of NALM6 cells treated with FHD-286 measured by FUCCI reporter cell lines. E) Level of disease in NSG mice (N=3/group) engrafted with human B-ALL cell line NALM6 treated with FHD-286 or vehicle x 7days. *p<0.05
B-lymphoblastic leukemia (B-ALL) is the second most common cause of cancer death in childhood. Additionally, among survivors, the therapies currently used to treat children with B-ALL result in multiple short- and long-term toxicities. Therefore, there remains an urgent, unmet need for novel therapies that can both effectively target B-ALL and do so with less toxicity than our antiquated battery of chemotherapeutic agents. The goal of this proposal is to define the effects of FHD-286, a novel, orally administered agent that inhibits the chromatin remodeling SWI/SNF complex, in pediatric B-ALL.
The rationale for our work stems from several novel observations. First, our initial work treating healthy mice with a small molecule inhibitor of the SWI/SNF complex revealed that this therapeutic strategy is well tolerated. This work also demonstrated that SWI/SNF inhibition results in transient reductions in the number of B-cells, the normal counterpart to malignant B-ALL cells. We then leveraged cell line CRISPR knock-out screen data and found that one of the major catalytic components of the SWI/SNF complex, SMARCA4, is essential for the survival of B-ALL cells. We have also generated preliminary data showing that several B-ALL cell lines have inhibited cell growth when treated with FHD-286. Additionally, our work in acute myeloid leukemia revealed that anti-leukemic response to SWI/SNF inhibition is driven by changes in chromatin and gene expression upon treatment. These observations have led us to hypothesize that FHD-286 is a well-tolerated new agent that will be effective alone and in combination for the treatment of B-ALL by rewiring of core regulatory transcription factor networks. We will test this hypothesis in three specific aims:
- Define the therapeutic effects of SWI/SNF inhibition across B-ALL subtypes.
- Devise rational combination therapies incorporating SWI/SNF inhibition for the treatment of B-ALL.
- Define molecular mechanism of response to SWI/SNF inhibition in B-ALL.
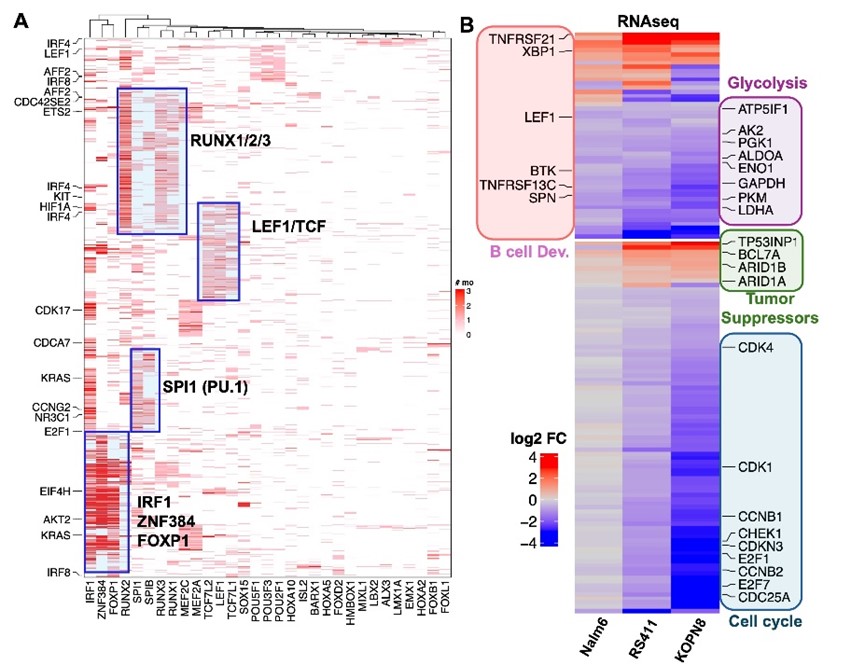
Figure 2: Chromatin and gene expression effects of FHD-286 on B-ALL cell lines. A) ATACseq of 3 B-ALL cell lines treated with FHD-286 x 48hr. Regions with lost accessibility include clusters of overlapping TF motifs. B) Gene expression changes with treatment with top categories of differentially expressed genes by gene ontogeny analysis with representative genes highlighted by colored boxes and text.
Long Read Sequencing to Characterize the Genome and Epigenome of Pediatric Leukemias
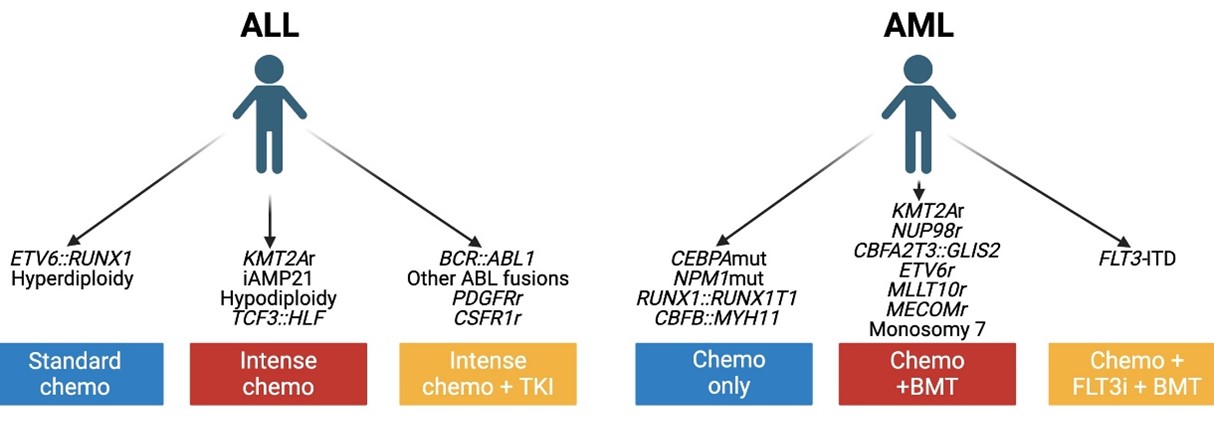
Figure 1: Current genomic risk stratification for leukemia patients.
The incorporation of genomic data into the pediatric leukemia care has improved outcomes by refining risk stratification, devising targeted therapies and individualizing drug dosing to minimize toxicity. Currently, to gather all needed genomic data, several assays are done in multiple labs in a process that is complex, slow, expensive and can result in treatment delays. Our goal is to revise genomic testing in pediatric leukemia.
| Assay | Information Generated | Turn-Around Time | |
|---|---|---|---|
| Tumor genomics | Fluorescent in situ hybridization (FISH) | Specific chromosomal translocations and aneuploidies | 3 to 5 days |
| Single nucleotide polymorphism (SNP) array | Chromosomal gains/losses | 1 to 2 weeks | |
| G-banding a metaphase chromosomes | Specific chromosomal translocations and aneuploidies | 2 to 4 weeks | |
| DNA mutation panel | DNA mutations | 2 to 4 weeks | |
| RNA fusion panel | Gene fusions | 2 to 6 weeks | |
| Germline genomics | TPMT genotype | Variants that influence 6MP metabolism | 1 to 2 weeks |
| NUDT15 genotype | Variants that influence 6MP metabolism | 1 to 2 weeks | |
| HLA typing | Immunotype for HSCT | 2 weeks |
Table 1: Current clinical genomic studies necessary for leukemia patients.
A new next-generation sequencing (NSG) modality called long-read sequencing (LRS) has several advantages over traditional sequencing. LRS interrogates long strands of DNA and RNA without fragmentation into small segments. The improved technology of LRS allows for the rapid detection of all the genomic variants currently identified by several different tests and even complex structural variants not readily identified by traditional methods. Additionally, a LRS assay called Fiber-seq, done concurrently with the standard LRS, detects epigenetic changes providing prognostically relevant information that has yet to be incorporated into routine care given a lack of available clinical assays.
We hypothesize that LRS can replace all existing clinical genomic tests, detect additional prognostic information, and thus enhance the speed and accuracy of risk stratification, therapeutic targeting and toxicity management in pediatric leukemia. By performing LRS on well-annotated diagnostic samples from pediatric leukemia patients, we will test this hypothesis in the following aims:
- Determine the accuracy with which LRS detects leukemia defining genomic lesions.
- Evaluate the capacity of LRS to detect germline genetic variants from diagnostic samples.
- Map the epigenome of pediatric leukemias using LRS and Fiber-seq.
By combining innovative LRS sequencing and epigenetic profiling technologies, our work will significantly advance diagnostic precision and treatment timeliness, ultimately significantly improving outcomes for patients with leukemia.
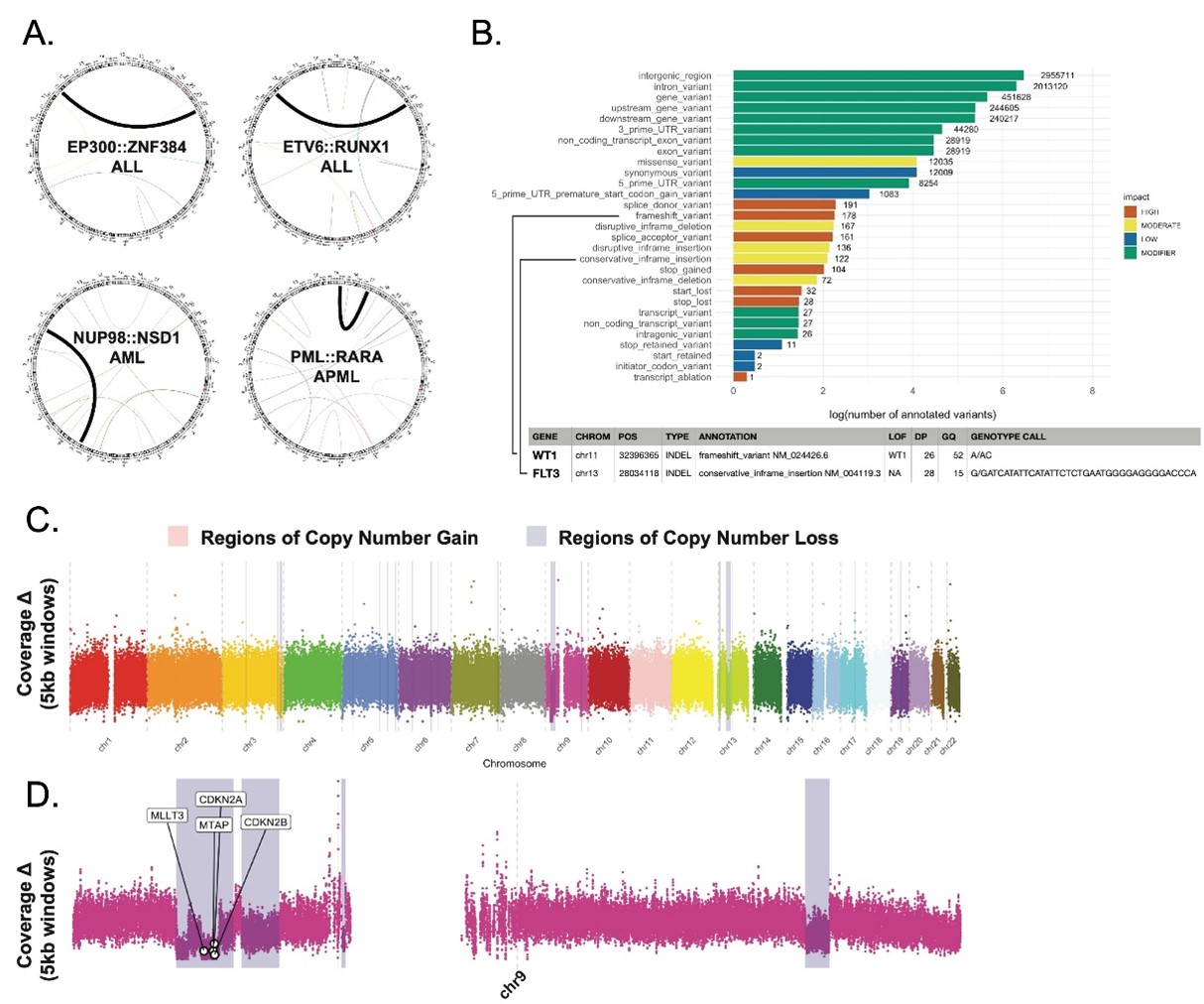
Figure 2: Accurate detection of leukemia-specific genomic lesions by LRS. A. All leukemia-defining fusions known by clinical testing were easily identified by LRS including ALL (top row) and AML/APML (bottom row) fusions. B. LRS identified genomic variants in an AML sample categorized by impact on protein coding. LRS identified the known WT1 and FLT3 mutations in this patient. C. Known copy number alterations across the genome. D. Zoom in on chromosome 9 showing known deletion of CDKN2A, CDKN2B and MLLT3.
Impact of Germline DNMT3A Mutations on Hematopoiesis and Cancer
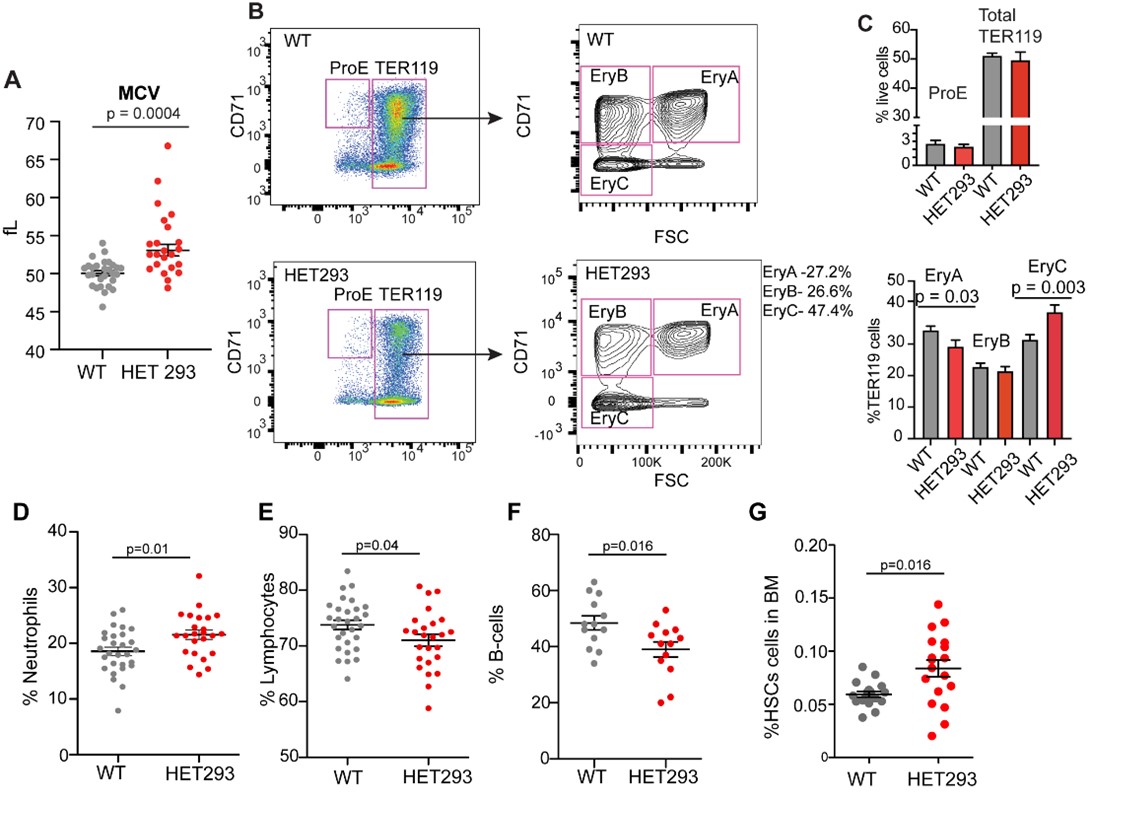
Figure 1: Hematopoietic perturbations in murine models of TBRS. Relative to wild-type (WT) littermate controls, mice with heterozygous germline deletions affecting amino acid 293 (HET293) have A) macrocytosis with B) evidence by flow cytometric analysis of the bone marrow (BM) to have altered maturation of erythroblasts with C) decreased percentages of maturing erythroblasts in the earliest stage (EryA) and increased percent in more mature the EryC stage. HET 293 mice are also characterized by D) increased percentages of blood leukocytes being comprised of neutrophils, E) reduced lymphocytes, due to F) reduced B-cells in the blood. G) HET293 mice also have increased percentages of LSK-SLAM hematopoietic stem cells (HSCs) in the BM.
Tatton-Brown-Rahman syndrome (TBRS) is an overgrowth disorder caused by germline heterozygous mutations in the DNA methyltransferase DNMT3A. DNMT3A is a critical regulator of hematopoietic stem cell (HSC) differentiation and somatic DNMT3A mutations are frequent in hematologic malignancies and clonal hematopoiesis. Yet, the impact of constitutive DNMT3A mutation on hematopoiesis in TBRS is undefined. Our work using multiple murine models of TBRS to:
- Determine the impact of specific Dnmt3a mutation on blood perturbations and leukemia risk over time.
- Define age-dependent impact of loss of DNMT3A on HSC function and hematologic phenotype.
- Examine the effects of age on human DNMT3A-mutant hematopoiesis.
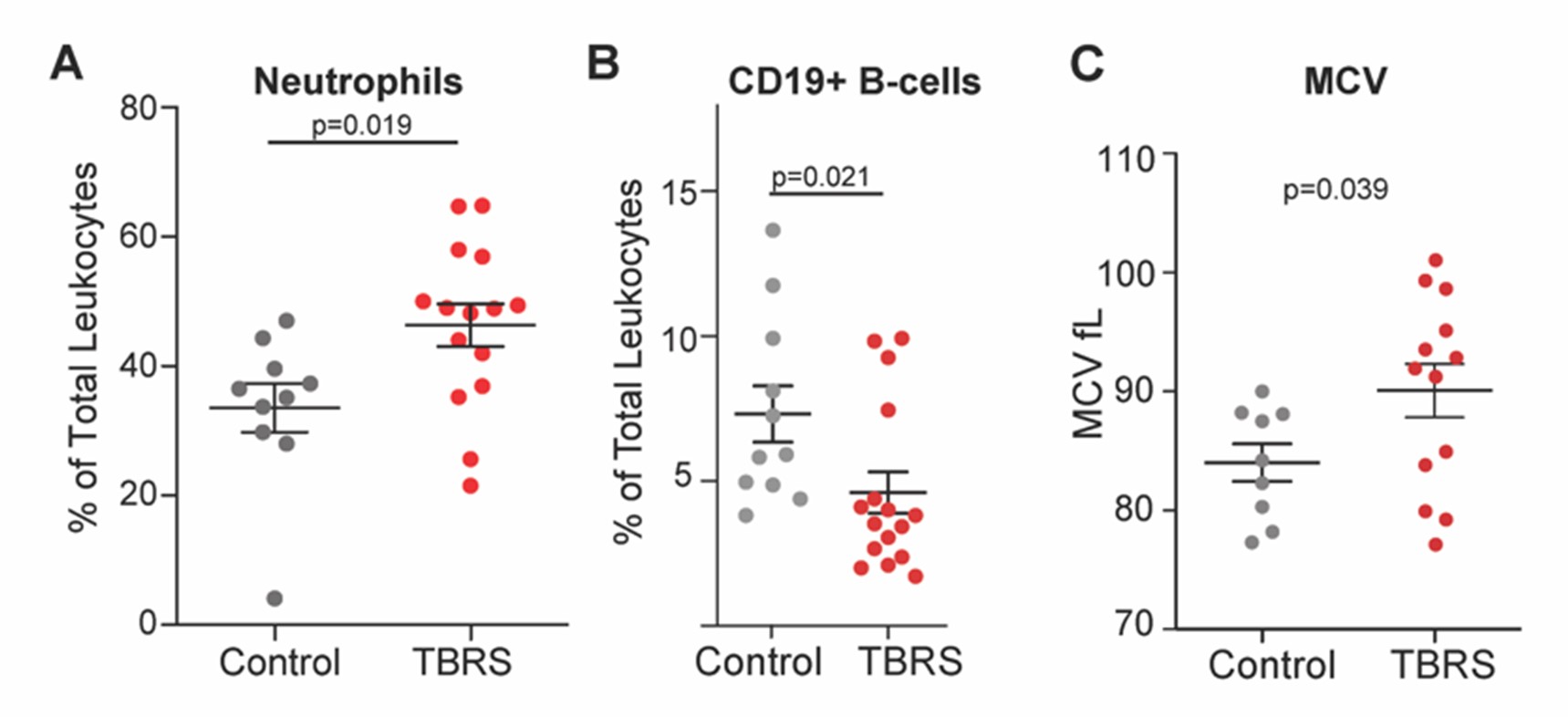
Figure 2: Hematologic perturbations in TBRS. Comparing individuals with TBRS to unaffected controls, TBRS is characterized by significantly A) increased neutrophils, B) reduced numbers of CD19+ B-cells and red blood cell macrocytosis as measured by mean corpuscular volume (MCV).
Use of High-Throughput Next Generation Sequencing of the Recombined Immunoglobulin Loci to Predict Relapse Risk in Pediatric ALL
This proposal will define novel applications of high-throughput sequencing (HTS) of the rearranged immunoglobulin loci (Ig) for improved risk stratification in pediatric B-lymphoblastic leukemia (B-ALL). B-ALL is the most common cancer in childhood and one of the leading causes of cancer death in children. Outcomes have improved over the last decades, but many patients still relapse, die of their disease, or suffer complications of therapy. Next generation Ig HTS is a DNA based assay for detection of minimal residual disease (MRD) in B-ALL, and work from our group and others has demonstrated that it is the most sensitive method of MRD detection available. In addition to the capacity of the assay to sensitively detect MRD, the composition of Ig sequences at diagnosis may be of biological and clinical relevance in and of itself. Yet, while Ig HTS is a proven technology, it has not been incorporated into the standard risk stratification algorithms for pediatric B-ALL. This substantial lag of clinical practice behind technological advancement is due to a lack of pediatric data on possible uses of Ig HTS in pediatric B-ALL.
Hypothesis and objectives: The central hypothesis of this proposal is that HTS of Ig loci in B-ALL can be applied prospectively to identify B-ALL subgroups with biological and prognostic relevance not defined by traditionally used risk predictors. To fill this knowledge gap, we will leverage data and samples from B-ALL patients enrolled on AALL1731, the largest therapeutic trial being run by the Children’s Oncology Group (COG).
Specific aims:
- Determine the significance of clonal Ig composition in pediatric B-ALL.
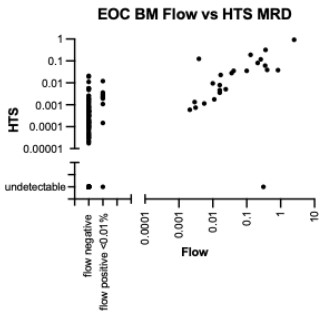
- Define the incidence and significance of discordance between flow cytometry determined MRD and HTS MRD.
- Comparative analysis of bone marrow MRD by HTS and flow cytometry at later therapy timepoints.
- To determine if Ig HTS on cell-free DNA from CSF can track CNS leukemia response to therapy and predict CNS relapse.
Significance: Capitalizing on our unique access to the COG AALL1731 clinical study, findings from this bench-to-bedside research may lead to the incorporation of Ig HTS into the routine risk stratification of pediatric B-ALL, identifying patients in need of different therapeutic approaches than is currently predicted.
Defining Genomic Prognosticators in Pediatric B-ALL in the Era of Blinatumomab
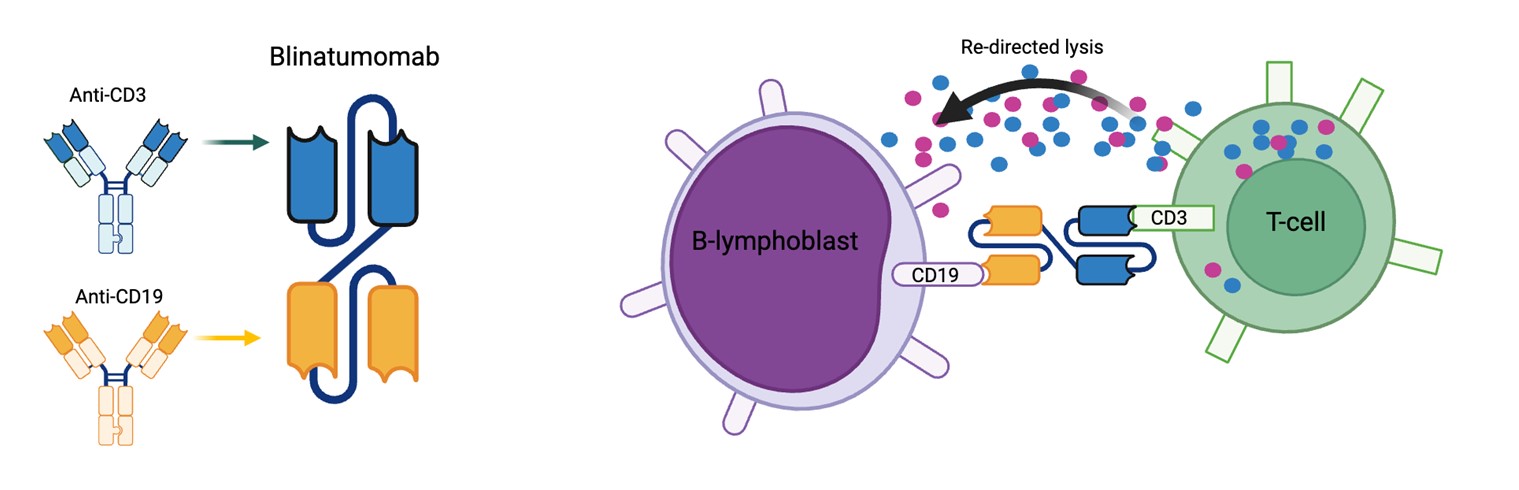
AALL1731 set a new treatment standard for children with B-ALL, in which the majority of such children will now be treated with chemotherapy and two non-sequential cycles of blinatumomab. Across the subgroups studied, chemotherapy plus blinatumomab resulted in outstanding outcomes previously seen in only the most favorable risk patients. Moving forward, a major goal of the COG ALL committee is to de-intensify therapy by removing elements of traditional chemotherapy, and as such minimize acute- and long-term side effects, while maintaining excellent outcomes.
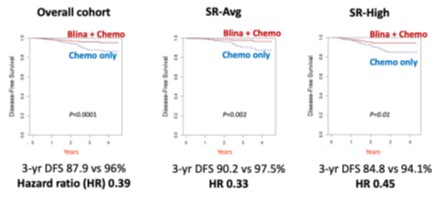
One significant challenge to this goal is our limited knowledge pertaining to risk of bone marrow relapse in the context of treatment including both chemotherapy and blinatumomab. The last several decades resulted in increasingly sophisticated knowledge in leukemia genetic predictors of relapse risk, from traditional cytogenetic lesions such as KMT2A rearrangements to more recent discoveries such as Ph-like gene expression and PAX5 alterations. Whether these and other genetic lesions hold similar prognostic value among patients treated with blinatumomab is unknown.
Bone marrow samples available from AALL1731 patients who did and did not experience bone marrow relapses represent a unique possibility to, for the first time, establish a genetic prognostication system for children with B-ALL receiving upfront chemotherapy and blinatumomab. We therefore propose to comprehensively characterize the genetic landscape of the AALL1731 randomized cohort using the well-established methods of whole genome sequencing (WGS) and whole transcriptome sequencing (RNA-seq) on banked diagnostic bone marrow or peripheral blood from the randomized cohort. The findings of this work will ensure that in successor COG clinical trials, deintensification of therapy is reserved for those patients who do not have any adverse genetic lesions discovered through these analyses.
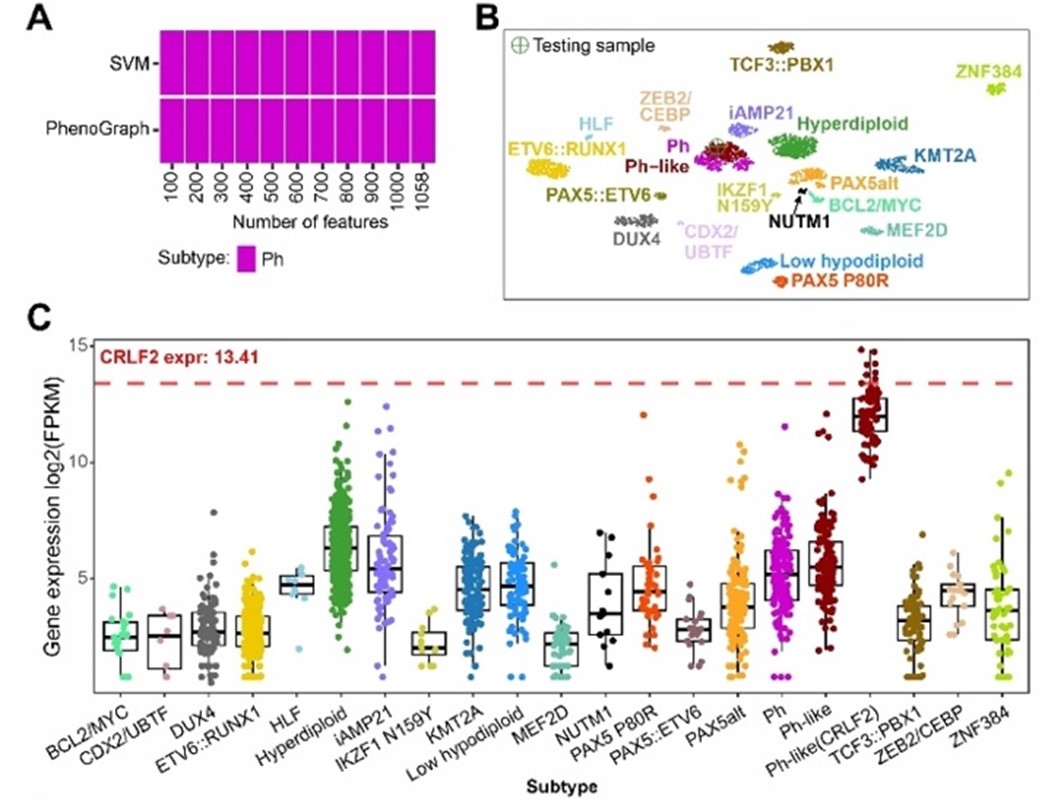
Figure 1: GEP-based B-ALL classification by MD-ALL. (A) Subtype prediction by SVM and PhenoGraph models using different numbers of feature genes. (B) UMAP projection of 1,058 feature genes shows the test sample clustering with the Ph/Ph-like group. (C) CRLF2 expression across B-ALL subtypes highlight overexpression in CRLF2r cases.
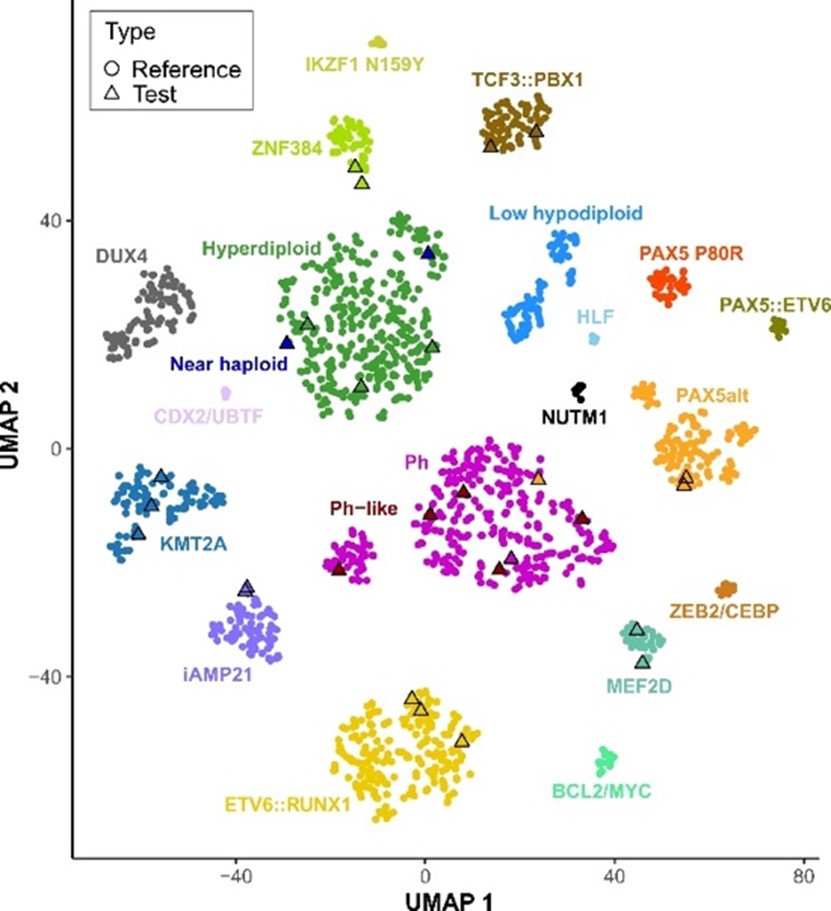
Figure 2: UMAP visualization of GEPs from 1,821 reference B-ALL samples (circles) and 28 test cases (triangles).